Research
Research
Organic
Our group seeks to apply state-of-the-art computational tools towards the efficient elucidation of mechanisms and factors that control the reactivity and selectivity of complex modern synthetic organic reactions. Unprecedented growth in new computational technologies and theories has made a vast spectrum of synthetic reactions amenable to computational analyses. The ultimate achievement is the complete understanding of the structural and energetic factors responsible for reactivity and selectivity. Our group and others have demonstrated that this is already reality.The same success is rare for chemical transformations where the structures are large, significant conformational flexibility is present, and/or the interactions responsible for the reactivity and selectivity are largely non-covalent and/or weak. Computational analyses of complex synthetic reactions often trail significantly behind experiments, while some are simply intractable. We address these challenges by harnessing software and hardware tools already in existence as well as developing our own.

Multi-Component Reactions
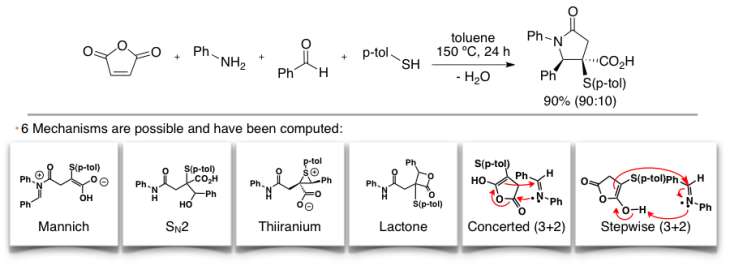
Multi-component reactions are particularly difficult to elucidate due to the large number of possible pathways. However, the implementation of our in-house automation techniques and computational tools has made this process feasible. We discovered, in collaboration with the Shaw Lab, the mechanism of the four-component synthesis of γ-lactams. The understanding of this reaction led us to investigate the origins of diastereoselectivity in the entire series of formal cycloaddition between cyanosuccinic anhydrides and imines.

Thiourea Catalysis
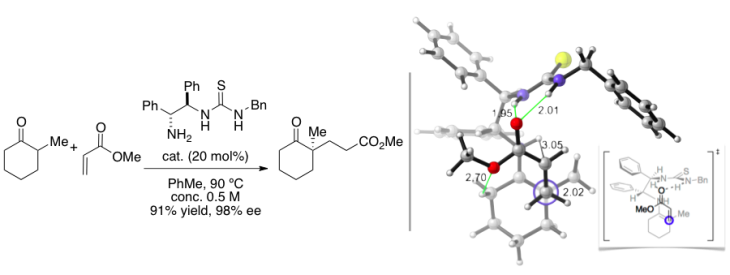
Our lab is investigating the mechanism and enantiomechanics of bifunctional thiourea organocatalysts for Michael reactions developed in the Carter lab here at OSU. Catalysis by these thiourea compounds is effected in two ways: 1) condensation of the catalyst's free amine onto the α-methyl cyclohexanone forms an enamine, activating the Michael acceptor; and 2) intramolecular hydrogen bonding with thiourea activates the electrophile's carbonyl.

N-Heterocyclic Carbene (NHC) Catalysis
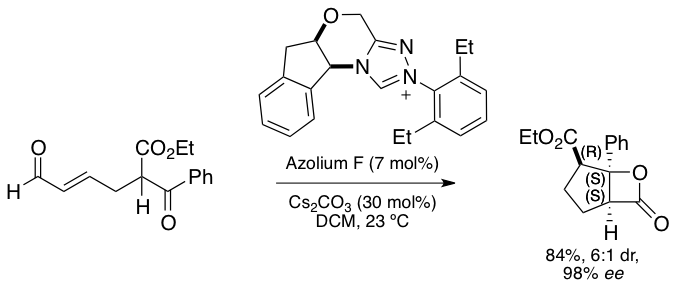
In a collaboration with the Scheidt lab at Northwestern University, our lab is investigating various aldol and homoaldol transformations catalyzed by NHC organocatalysts. In this NHC-catalyzed dynamic kinetic resolution, β-ketoesters are asymmetrically transformed to highly substituted β-lactones (Angew. Chem., Int. Ed.2012, 51, 7309–7313). Computational work by our lab has discovered both a deviation from the originally proposed mechanism and the origins of diastereoselectivity.

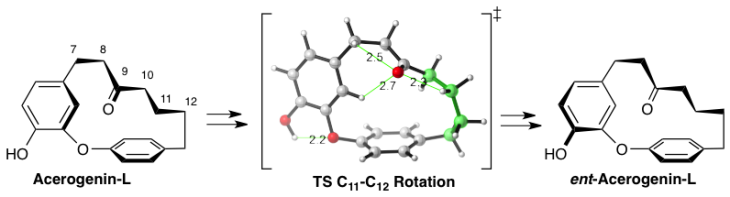
Conformational Chirality of DAEH
Diarylether heptanoids (DAEH) natural products do not have stereocenters but can manifest conformational chirality. However, the ability to predict conformational chirality in these compounds had not been studied previously. In collaboration with the Beaudry Lab here at OSU, we have elucidated the structural features and energetic details related to the stereoisomerizations of four representative DAEHs. We discovered a general way to predict the existence of conformational chirality in any arbitrary DAEH.
Materials
Our group participates in materials science research as part of the Center for Sustainable Materials Chemistry (CSMC). We contribute computational and theoretical expertise to the CSMC research mission, providing a fundamental understanding of aqueous metal hydroxide clusters and translating these discoveries to guide experimental efforts by research groups within the CSMC and beyond. While research in the PHYC group is computational, the ultimate goal is the complete understanding of these metal hydroxide clusters such that computations are no longer necessary to predict experimental outcomes or properties of these clusters.
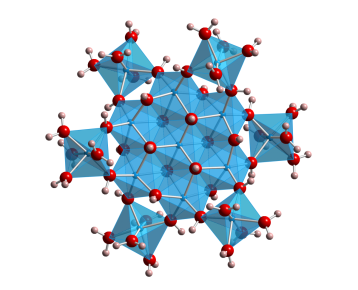
Metal Oxide & Hydroxide Clusters
As part of the Center for Sustainable Materials Chemistry (CSMC), our group examines the stabilities and various properties of metal oxide and hydroxide clusters. Primarily, these studies focus on the metal oxide and hydroxide clusters of Group 13 cations (Al, Ga, and In). The "flat" tridecamer structure of these Group 13 metal clusters is central to our research. Aqueous solutions of these clusters can be used to form high quality amorphous thin films for applications in electronics. Additionally, metal hydroxide cluster chemistry is of interest to environmental scientists and geochemists. Our current research explores the thermodynamic stabilities of these clusters in solution to better understand the factors that give rise to specific clusters over others. Additionally, we are studying of the electronic structure of these clusters, and seeking high accuracy methods for the prediction of these properties.
Spectroscopic Analysis
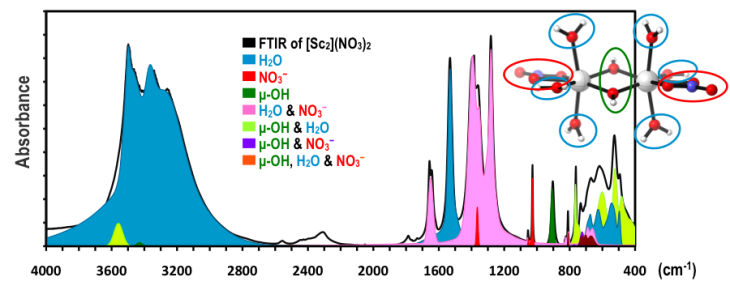
The experimental and computed IR spectrum of [Sc2(OH)2(H2O)6(NO3)2](NO3)2. The IR contributions of each functional group are also shown.
Spectroscopic analysis can assist experimentalists in examining new classes of materials with classic forms of spectroscopy, most commonly with Raman and IR spectrscopy. Our lab has developed a computational strategy to identify the exact identities of metal hydroxide clusters present in an aqueous solution environment; this complements experimental spectroscopic methods. Our strategy identifies the signature peaks of a given cluster and quantifies the contributions of each vibrational mode in the IR, Raman, and NMR spectra. Published paper can be found here.
Amorphous Metal Oxides
In collaboration with Corning, Inc., our group has developed a method to generate amorphous metal oxides. By definition amorphous solids have no repeatable structure and therefore the atomistic detail cannot be elucidated with experiments. This is where computational chemistry can bridge the gap. Once we generate an atomistic model of the amorphous metal oxide we can compute any physical and electronic properties. These computations can then assist experimentalists in their search for novel materials with their properties of interest. Due to specific electronic and physical properties, the InGaZnO4 (IGZO) family of amorphous metal oxides is of academic and industrial importance. By generating different stoichiometric compositions of IGZO we are able to predict these electronic and physical properties. Our aim is to find novel amorphous materials that will become the cornerstone in the next generation of electronic devices.
Bioorganic
Catalysis based on the fundamentals of natural enzymes and proteins is the new frontier of green and efficient synthesis. Enzymatic acceleration ranges from 105 in Cyclophilin to 1017 in Orotidine monophosphate decarboxylases (“A proficient enzyme.” Science1995, 267, 90–93)The dramatic miniaturization of nature’s enzymes into catalytic peptides is a significant achievement in modern synthetic chemistry and biology. Structural preorganization in the transition states is critical for imbuing reactivity and selectivity (“Asymmetric Catalysis Mediated by Synthetic Peptides.” Chem. Rev.2007, 107, 5759–5812). However, these factors are poorly understood for peptide catalysis due to the large size, conformational flexibility, and weakness of the interactions responsible for preorganization and transition state stabilization. By quantifying these factors using our strategies and tools, we will begin to create theories that govern the selectivities of these reactions and eventually contribute to the rational design of peptide catalysts.
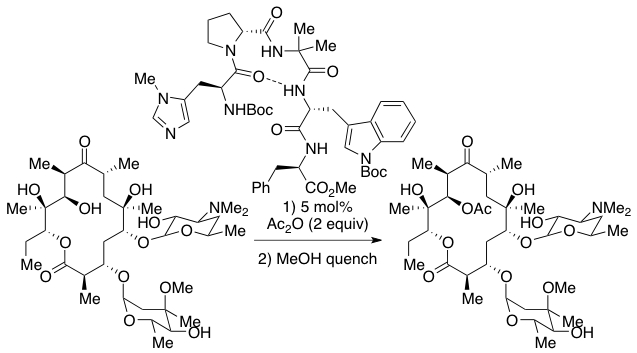
Tetrapeptide ((Π-methyl)His-Pro-AIB-Phe) Catalyzed Acylation Reaction
The PHYC lab is studying the mechanism and origins of selectivity of the asymmetric acylation of 2-hydroxycyclohexyl acetamide. Experiments show that the D-proline catalyst epimer affords the 28:1 enantioselective preference for (R,R)-2-acetamidocyclohexyl acetate ("A Biomimetic Approach to Asymmetric Acyl Transfer Catalysis” J. Am. Chem. Soc. 1999, 121, 11638-11643").
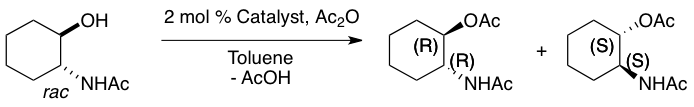
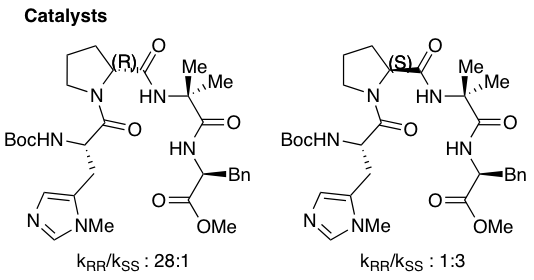
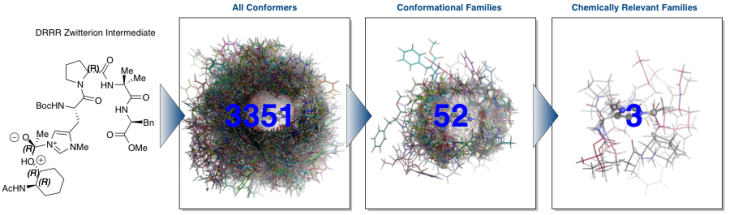
The enantio-determining step (below) involves the nucleophilic attack of the acyl-catalyst by the substrate. This substrate-catalyst complex is inherently flexible and hence, has a large conformational space. The PHYC lab has developed a cluster-conformational protocol that takes advantage of multiple computational programs to discover the chemically meaningful families from such complex systems. As shown below, we have discovered that of the 3351 possible conformers, 52 families exist, and only 3 of these are chemically relevant. This optimization process makes this reaction, otherwise computationally intractible, feasible to elucidate through extensive quantum mechanical methods.